
Total aligned bases
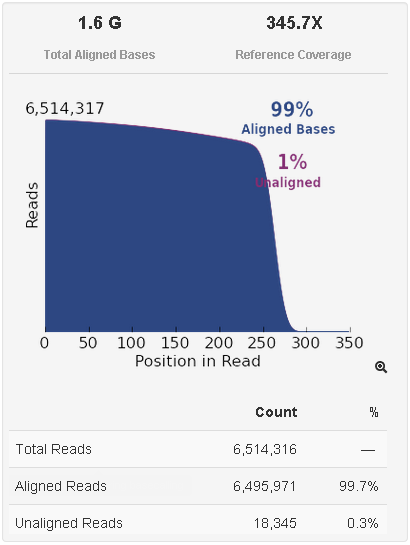
The following table describes metrics in the Total Aligned bases area.
|
Metric |
Description |
|---|---|
|
Total Aligned Bases |
Number of filtered and trimmed aligned base pairs reported in the output BAM file. Total number of bases aligned to the reference sequence. Excludes the library key, barcodes, and 3' adapter sequences. |
|
The average of the number of reads that cover each reference position: total aligned bases divided by the number of bases in the reference sequence. Does not consider enrichment. |
|
|
Percentage of Total Aligned Bases out of all reads. |
|
|
Percentage of bases not aligned to references. |
|
|
Total Reads |
Number of reads generated during basecalling. |
|
Number of reads that aligned to the reference genome. |
|
|
Number of reads that did not align to the reference genome. |
The graph in the Total Aligned reads column plots number of aligned (in blue) and unaligned (in purple) bases byposition in an aligned sequence. (The purple area cannot be seen easily when it is under 3 or 4 percent.)
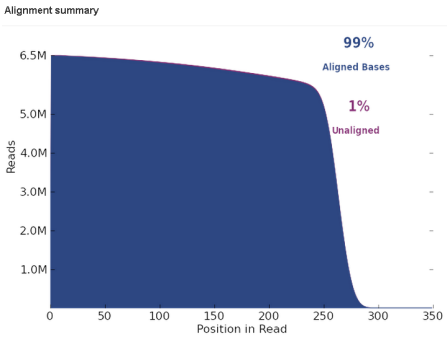
For each position in an aligned sequence, the height of the blue area shows the number of aligned bases at that position. The purple area shows the number of unaligned bases at that position. Unaligned bases are not shown by the absolute height on the number of bases axis, but by the difference between the purple height and the blue height.