
Variant Calls by Allele table
The following list summarizes the features of the Variant Calls table:
-
Each position is a link to open the variant in IGV. In some browsers, you save the igv.jnlp file to your local system, and then click on igv.jnlp to open the IGV browser.
-
You can export selected variants to a table file or to the Life Technologies PCR and Sanger Sequencing For TaqMan® Assay Design web sites.
-
Click on a column header to order the table by the contents of that column.
-
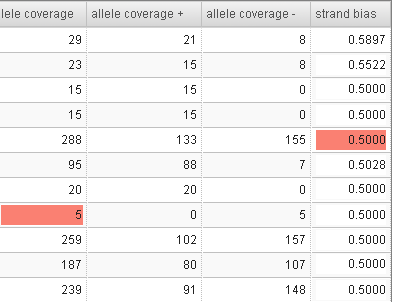
For candidates that are filtered out, the filtering reason is highlighted in the table. For example:
The main columns are described in the following table. Use the View tabs on the right of the table to change the display of the columns on the right:

|
Position |
The chromosome (or contig) name in the reference genome, and the one-based position in the reference genome. |
|
Ref |
The reference base(s). |
|
Variant |
Variant allele base(s). |
|
Var Freq |
Frequency of the variant allele. |
|
Quality |
Phred-scored quality field. Larger values mean more certainty in the call. Typically very large for reads strongly distinguishing variants (SNPs) with good depth; that is, under the model assumed, evidence is overwhelming for the variant or for the reference. Marginal values in this field can mean either the reads do not distinguish the variant well or there is insufficient depth to resolve, or the observed allele frequency is near the cutoff. Filters to compensate for the cases in which the model assumptions are not true are found in the INFO tags. Computed by posterior probability that the sample variant allele frequency is greater than the min-allele-frequency specified for the variant type (if a variant), or posterior probability that the variant allele frequency is below this threshold (if a reference call). Posterior probability computed conditional on the reads observed, includes sampling variability. |